One morning during my senior year in college, I was driving my car around town and had the sneaky suspicion that my car's lighter wasn't working. I pushed the lighter in for a couple of minutes and then examined the end. Since the coils weren't red it only fueled my suspicion. What I did next stands firmly in the pantheon of stupid things I've done in my life...I placed my thumb over the coils to see if they were hot. I honestly can't tell you why I did this, but every sensation for the next couple of seconds has been burned into my memory. It took about four months for my thumbprint to grow back. Long story short, just because you can do something doesn't mean you should.
Molecular evolutionary analyses have gotten extremely easy to perform in the last couple of decades. There exists a variety of plug and play and freely accessible computational tools and programs that will chew up any DNA sequences you input and spit out numerical results related to evolutionary scenarios. These programs will work even if you input completely fake sequences and made up taxa. Like many programs (looking at you t-test in Excel) they are agnostic to the underlying assumptions. It's up to the user to make sure comparisons are valid.
dN/dS and it's cousin Ka/Ks are often used to measure whether positive selection has acted on a set of nucleotide sequences. Explained very briefly, for a given nucleotide sequence, dN is the number of non-synonymous nucleotide changes in the focal sequence compared to a reference sequence. dS is the number of synonymous changes in those same sequences. For these analyses, synonymous mutations are assumed to be selectively neutral and thus represent the background rate of evolution. If there is no selection acting on a protein sequence, it is assumed that non-synonymous changes should occur as frequently as synonymous. Therefore, all else equal (in a very unrealistic world), proteins under no selective pressure should have a dN/dS ratio around 1. A dN/dS ratio > 1 indicates positive selection because in this case non-synonymous changes occur more frequently than the baseline synonymous rate and selection must therefore be increasing dN. A dN/dS ratio < 1 indicates purifying selection because non-synonymous changes are being selected against and are therefore found in lower numbers than expected. I'm glossing over a lot of nuance in these analyses but that's the 30,000 foot view.
While you can calculate dN/dS for a bunch of sequence blind to their evolutionary relationships, this number doesn't really give you much insight into the evolutionary process. Alternatively, if you have a set of sequences for comparison and a phylogenetic tree showing evolutionary relationships between these sequences you can orient where and at what rates these nucleotide changes have occurred. If you use a program uses statistics to infer evolutionary models (like PAML), you will be asked to input a phylogeny for this very reason. To avoid confounding yourself with circular calculations, it's important to use phylogenies estimated from loci outside the scope of the analysis, even though in many cases phylogenies built from your loci of interest will (for the most part) match those built from the rest of the genome.
Genes undergoing horizontal gene transfer (by definition) have evolutionary histories and phylogenies that differ from the rest of the genome. You can certainly build a phylogeny using these sequences, and you can use this as your tree for calculation of evolutionary rates, and PAML will accept your input files without questioning them. You will be given a numerical answer when calculating dN/dS (or more specifically omega). In the best case scenario you will then go on and publish papers, generate press releases, and maybe even sit in on a podcast to describe your awesome results. The problem that arises though, is you have no clue as to the true evolutionary relationships between the genes of interest. The phylogeny just tells you how similar a given set of sequences are, but it doesn't necessarily tell you the evolutionary history of horizontally transferred genes. Take for instance a specific gene present on a highly transmissible plasmid (Fig. 1).
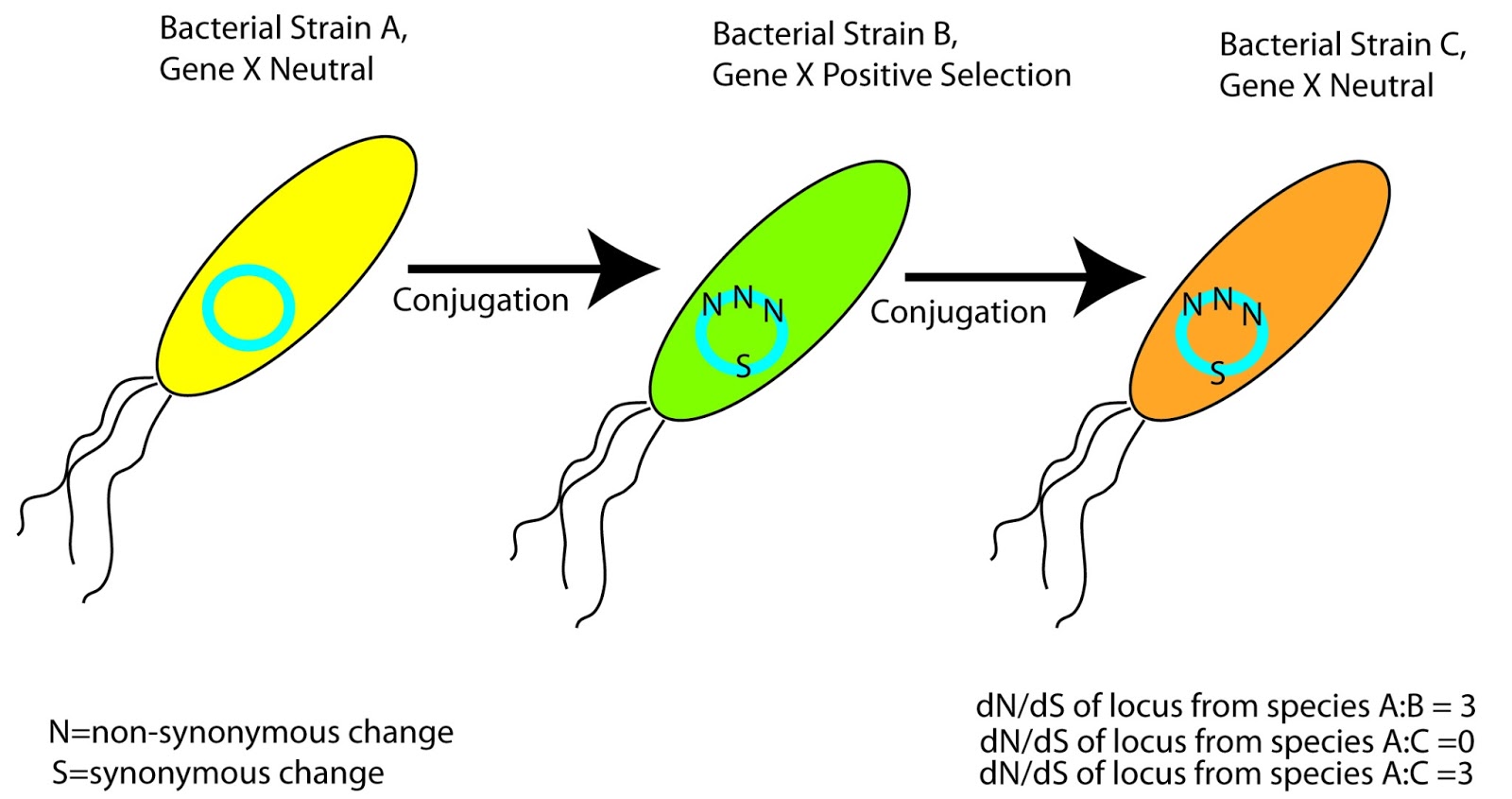
Figure 1: If Your Gene of Interest Undergoes Horizontal Gene Transfer, Good Luck Guessing It's Evolutionary History
This plasmid can be passed around from strain to strain, and may experience a number of different environments. In some environments/backgrounds your gene of interest may be under positive selection, in some cases negative, in others no selection at all. Unless you know the complete transmission history of this plasmid you are stuck sampling gene sequences from the last strain it's found in. In the example in Fig. 1, you might assume that your gene of interest is under positive selection in Species C compared to A but in reality it's neutral in both cases. Moreover, if you were to compare this gene from species B and C you would think that there was no difference in selection between the two. When you build a phylogeny with your genes of interest, and sit down to calculate dN/dS (or even worse omega), all you can see is that non-synonymous changes have occurred at some point since the divergence point between two sequences.
Alternatively, bacterial loci may recombine with divergent orthologues from closely or distantly related strains through processes like natural transformation. In this case only a small fraction of the codons in a gene may be replaced with divergent sequences, but such events can totally skew dN/dS ratios (Fig. 2).

Figure 2: Site Specific Recombination Disrupts Interpretation of dN/dS
Although the neutral theory provides a decent null hypothesis for understanding selection on mutational events in natural populations, there is no comparable model for bacterial recombination. Are a majority of homologous recombination events neutral? Are a majority of recombination events subject to positive selection? We have very little understanding of how selection and homologous recombination interact in natural bacterial populations, but we do know that recombination is very frequent within some lineages even across "housekeeping" genes. Along these lines, a recent study investigating Staphlococcus aureus and Clostridium difficile suggests that frequent recombination events bring with them many more synonymous polymorphisms than non-synonymous. Offhand, I can think of many scenarios where dN/dS ratios wouldn't match the underlying evolutionary dynamics because of horizontal transfer but the fact of the matter is that we as a community have no clue as to what to expect.
All this brings me to the ultimate take home message of this post. If there's a hint that horizontal gene transfer may be affecting your analyses of selection, probably best to avoid having me as a reviewer because it's going to take a heck of a lot to convince me that the methods are sound.
Update: Matt Barber (@MattFBarber) reminded me of a great review paper from Jesse Shapiro relevant to this post, but which goes much more in depth into a variety of analyses. Also came across an awesome recent paper from Sheppard et al. (h/t Mark Pallen, who referrs to the "et al." as the Sheppard, Didelot, Falush posse) on how to use bacterial recombination to your advantage for GWAS studies.