
In addition to Illumina data for the reference parent strain, I also received sequencing reads for this brown colony phenotype strain as well as a couple of other independent derivatives of strain 28a24 (which will also be useful in this mutation hunt). There are a variety of programs to align reads to the draft genome assembly, like Bowtie2 and Maq. However, because it's a bit tricky to align reads back to draft genomes and because I'd like to quickly and visually be able to inspect the alignments, I'm going to use a program called Geneious for this post.
The first step is to import the draft assembly into Geneious, and in this case I've collapsed all of the contigs into one fasta read so that they are separated by 100 N's (that way I can tell what contigs I'm artificially collapsing from real scaffolded contigs). Next I import both trimmed Illumina paired read files, and perform a reference assembly vs. the draft genome. In Geneious it's pretty easy to extract the relevant variant information, like places where coverage is 0 (indicating a deletion) or small variants like single nucleotides or insertions/deletions. Here's what the output looks like:
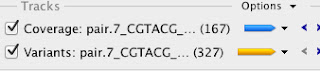
For the reads from the brown phenotype strain there are basically 167 regions where there are no reads mapped back to the draft genome. A quick bit of further inspection shows that these are all just places where I inserted the 100 N's to link together contigs. There are also 327 smaller variants that are backed up with sufficient coverage levels. My arbitrary threshold here was 10 reads per variant, but my coverage levels are way over that across the board (between 70 and 100x).
Here is where those other independent derivatives of the parent strain come into play. There are inevitably going to be assembly errors in the draft genome, and there are going to be places where reads are improperly mapped back to the draft genome. Aligning the independent non-brown isolate reads back against the draft genome and comparing lists of variants allows me to cull the list of variants I need to look more deeply at by disregarding variants shared by both. After this step I'm only left with 3 changes in the brown strain vs. the reference strain.
The first change is at position 2,184,445 in the draft genome, but remember that the number here is arbitrary because I've linked everything together. This variant is a deletion of a G in the brown genome.

Next step is to extract ~1000bp from around the variant and use blastx to give me an idea of what this protein is.


The last variant is the most interesting. It's a T->G transversion in a gene that codes for homogentisate 1,2-dioxygenase (hgmA)


To illustrate the function of this gene, I'm going to pull up the tyrosine metabolism pathway from the KEGG server (hgmA is highlighted in red).

The function of HgmA is to convert homogentisate to 4-Maleyl acetoacetate. Innocuous enough and I'm no biochemist so in pre-Internet world I'd be somewhat lost right now. Luckily I have the power of google and knowledge of the brown phenotype so voila (LMGTFY). Apparently brown pigment accumulation in a wide variety of bacteria is due to a build up of homogentisic acid. There is even this paper in Pseudomonas putida. Oxidation of these compounds leads to quinoid derivatives, which spontaneously polymerize to yield melanin like things. Without doing any more genetics I'm pretty sure this is what I've been looking for. It's a glutamate to an aspartate change at position 338 in the protein sequence, a fairly innocuous change but which is (if this truly is the causal variant) in a very important part of the protein sequence. From here, if I were interested, I would clone the wild type version of hgmA and naturally transform it back into the brown variant of 28a24 to complement the mutation and demonstrate direct causality. I might also try and set up media without tyrosine to see if this strain is auxotrophic, which is a quality of other brown variants (see the P. putida paper above). For now I'll just leave it at that and move on to another interesting project that I can blog about.
No comments:
Post a Comment