Ahhh...it's grad/med/dental/etc school application season. For the first time ever I'm sitting down to write multiple letters of recommendation for former students and researchers in my lab. I've now seen the process from all sides: as a prospective undergraduate researcher, as a tenure track PI asked to write letters, and as a grad school admissions committee member. I remember being horribly blindsided by the process as an undergrad (they require 3 letters from DIFFERENT people?) and hope to offer a couple of words of advice for those prospective students.
1. Email me way before the deadline to ask about writing a letter. There is nothing worse than finding out that I have to pop out a letter of recommendation in the next couple of days. My life as a PI is stressful enough that I only survive by making lists of things to get done over the next couple of weeks. If you wait until the last minute, even if I think you have a great future as a post-grad, there is a significant chance I will decline because there aren't enough minutes in the day.
2. If you are a student in my class, don't wait until after the final to introduce yourself. I'm pretty good at remembering names and faces and know who participates in class and who doesn't. If you are ultimately thinking about grad school it's a good idea to participate in discussions in my class and answer questions. Not only does this give me ammo to put in the letter, but it makes a good first impression (which never hurts).
3. Get a good grade in my class. I usually only write letters for students that get A's in my class. I will make exceptions if you are an active participant (see #2) or if you've made a good hearted effort to improve your grade as the semester went on. This doesn't just mean doing better on tests, but it means coming to office hours and showing an interest in the material. I can't say this enough, enthusiasm is one of the greatest assets for prospective students.
4. Help me out. A letter of recommendation is just that...I am writing to back you in your pursuit of higher education. You can help by scheduling meetings to come and talk with me in person, give me a feel for what your interests/goals are. Let me know why you want to go to grad school. Tell me stories that illustrate why I should give you my seal of approval. A letter can be pretty dry if all I can write is that you got an A in my class. Seriously, help a brother out here, it will go a long way towards making your letter the best it can be.
5. Don't make me search for addresses to email the letter to or websites to log in to. Please, give me the links and I promise you that it will get done much faster and more smoothly.
6. Ask if there are spots for undergraduate researchers in my lab. There is nothing better in a letter of recommendation for grad school than positive comments about a student's laboratory skills and dedication. All grad school is is trying to figure out how to make experiments work, and you've got a good head start if you've already had this experience. Obviously, the earlier you do this the better.
7. Remember, I'm doing this to help you. There is nothing in my contract that requires me to write letters of recommendation. Seriously, this is a favor I'm doing for you.
Have to write letters? Check this out.
Any other thoughts? Feel free to contribute in the comments.
Monday, November 26, 2012
Thursday, November 15, 2012
Co-corresponding authors
I was involved in a bit of a discussion over twitter this morning over the value of co-corresponding authors on manuscripts. This has inspired a Drugmonkey blog post with some good comments. Because I've actually published a paper with co-corresponding authors (here), I thought that I could provide a slightly different insight into the process.
First off, what does it mean to be a corresponding author? Traditionally, the corresponding author spot on a paper is there in case researchers stumble across your manuscript and have questions or requests for reagents. This has morphed into somewhat of a status symbol with the increase in number of authors on papers because corresponding authors are seen as having "ownership" (for lack of a better word) over the published project. For instance, in the CV for my tenure packet I list where I am the corresponding author on manuscripts from my own lab that my postdoctoral advisor is also an author on. Maybe this matters, maybe it doesn't, but I see it as a way to point out projects that I have taken more of a lead role on.
So after that brief intro, here's my experience with co-corresponding authorship. Back in 2009 we published a paper on sequencing and assembly of a Pseudomonas syringae strain (here). This paper included biological data as well as a computational pipeline to that we used to assemble the genome. My postdoctoral advisor, Jeff Dangl, was the sole corresponding author on this paper. Jeff is an incredible biologist, but is not the best programmer in the world. Dangl was the perfect corresponding author for any biological question (strain/construct requests, etc...) from that paper. However, Jeff would get emailed questions concerning the computational pipeline and inevitably would forward the emails to the people (Corbin Jones and me) that could actually answer them. This was a bit frustrating.
To prevent this situation in our next paper in this series, which expanded this pipeline and analyses across 19 strains, both Corbin and Dangl were corresponding authors with a note that Jeff would handle the biology and Corbin would handle the computational questions.
One important thing to note...in each case the lead author has been the one actually formatting and uploading the paper to the journal, and also dealt with actual correspondance to the editor of the journal after submission without being listed as corresponding author. As many know, those are the most fun and fulfilling parts of manuscript submission...
First off, what does it mean to be a corresponding author? Traditionally, the corresponding author spot on a paper is there in case researchers stumble across your manuscript and have questions or requests for reagents. This has morphed into somewhat of a status symbol with the increase in number of authors on papers because corresponding authors are seen as having "ownership" (for lack of a better word) over the published project. For instance, in the CV for my tenure packet I list where I am the corresponding author on manuscripts from my own lab that my postdoctoral advisor is also an author on. Maybe this matters, maybe it doesn't, but I see it as a way to point out projects that I have taken more of a lead role on.
So after that brief intro, here's my experience with co-corresponding authorship. Back in 2009 we published a paper on sequencing and assembly of a Pseudomonas syringae strain (here). This paper included biological data as well as a computational pipeline to that we used to assemble the genome. My postdoctoral advisor, Jeff Dangl, was the sole corresponding author on this paper. Jeff is an incredible biologist, but is not the best programmer in the world. Dangl was the perfect corresponding author for any biological question (strain/construct requests, etc...) from that paper. However, Jeff would get emailed questions concerning the computational pipeline and inevitably would forward the emails to the people (Corbin Jones and me) that could actually answer them. This was a bit frustrating.
To prevent this situation in our next paper in this series, which expanded this pipeline and analyses across 19 strains, both Corbin and Dangl were corresponding authors with a note that Jeff would handle the biology and Corbin would handle the computational questions.
One important thing to note...in each case the lead author has been the one actually formatting and uploading the paper to the journal, and also dealt with actual correspondance to the editor of the journal after submission without being listed as corresponding author. As many know, those are the most fun and fulfilling parts of manuscript submission...
Monday, September 24, 2012
Chasing Down the Cause of Random Experimental Results
I'm pretty sure that every wet lab biologist has a story or two or many about experimental controls behaving weirdly or in seemingly unexplainable ways. A very small percentage of the time, chasing down the underlying cause can actually lead to a Nobel prize. Much more often the underlying cause is unremarkable ("Oh, that was a ug instead of mg?"). We had one of these results in the lab last week and, while interesting and actually a real phenomenon rather than facepalmish error, it's still fairly unremarkable so I figure I'd share it here.
We are currently trying to measure mutation rates to rifampicin resistance in Pseudomonas stutzeri. We already have encouraging results showing how a particular genotypic change increases mutation rates to streptomycin resistance and to goal is to calculate mutation rates an additional phenotype so we could begin to think that the genotypic change generally increases mutation rates. For other reasons, we wanted to measure the mutation rates to rifampicin resistance in a streptomycin resistant background strain.
Since P. stutzeri is competent for natural transformation, we transformed a rif and strep sensitive strain (rifS and strepS hereafter) using genomic DNA from a rif and strep resistant strain (rifR and strepR). No problem yet as we got plenty of strepR colonies back.
The next step is to use a fluctuation test to calculate mutation rates to rifR using cultures started from a single strepR colony. The basic idea of a fluctuation test is to grow many independent cultures starting from very low cell densities (typically ~1000 cells, in order to insure that there are no rifR colonies at the start), and then plate the entirety of these cultures under selective conditions once the cultures have grown to appreciable densities. If there are no rifR cells at the beginning of growth , you can use the distribution of rifR colonies that appear across the independent cultures in order to calculate mutation rates.
The picture below describes what you might expect from a typical fluctuation test.

When you plate out samples at time 0, there should be no rifR colonies (which I'm showing as blue circles if present). After growth, in this case 24 hours, rifR cells will have arisen by spontaneous mutation INDEPENDENTLY in each culture. As you can see from the picture, the expectation is that some cultures will not contain any rifR cells, some will have a few, and some will contain many rifR colonies. Those with many colonies are called "jackpots"and may appear as confluent lawns, hence the totality of blue in the picture. There is a distribution of rifR cells across independent cultures because mutations that lead to rifampicin resistance will occur at different points of the growth curve in each culture. A single mutation that arises during the first cell division after starting the experiment will lead to jackpots because these rifR cells have the chance to divide many times before plating. A rifR cell that arises during the last division before plating will only be represented by one colony because it hasn't had the time or resources to divide and proliferate. You can use this distribution to actually figure out mutation rates. Here is Stan Maloy's explanation of the fluctuation test.
Back to our P. stutzeri experiments...when we plated out the fluctuation test for our (what we thought) strepR rifS isolate, we got this result back from the time zero plate.

There was a lawn of rifR bacteria, which is statistically higher than zero (not really, but go with me on this). Even though the mutation rate to rifR is relatively high, something was fishy here because there should have been no colonies. We went back to the original plate of strepR transformants, and streaked additional isolates to both strep and rif plates. Even though the strain which we had originally transformed was rifS, I was surprised when roughly half of the strepR transformants were also rifR.
What could explain this result? The mutations that give rise to the strepR phenotype usually occur within a gene called rpsL. This gene codes for one of the many proteins that make up bacterial ribosomes, and indeed, the mechanism of action for streptomycin is inhibition of translation. The first thing I did was investigate the genomic context, what genes surround rpsL, within the one previously sequenced P. stutzeri genome. Here's what I found at the Pseudomonas genome database (which is extremely handy BTW).

All annotated genes in this section of the P. stutzeri genome are represented as red boxes. rpsL is the red box surrounded by a very dark line. The numbers on the bottom of the picture display the position of each gene in the P. stutzeri genome (rpsL is roughly at position 888,000 out of ~4,000,000). Here's where it gets interesting. Mutations that lead to rifampicin resistance typically occur in a gene called rpoB. This codes for a subunit of RNA polymerase, which makes sense (again) because the mechanism of action for rifampicin is to inhibit transcription. As I described above, we originally made the strepR strain by transforming a rifS strepS strain with genomic DNA from a rifR strepR strain. rpoB and rpsL are only about 5000 bp apart and I originally had no clue that rpoB and rpsL were genomic neighbors. Since, during natural transformation, genomic fragments sized 5Kb and above can be recombined into the recipient genome I'm guessing that a substantial fraction of the cells recombined both strepR and rifR mutations during this step from the donor genomic DNA. Since the exact positions of recombination are essentially random, cells that remained rifS after transformation must have recombined smaller fragments that only contained the strepR mutation. Here's a quick picture of what I think happened.

Rifampicin resistance is colored red, streptomycin resistance is colored blue. Cells that are rifR strepR are colored purple. While there were likely rifR strepS cells generated after transformation, these won't grow on the strepR plate so that's why there aren't red colonies. Mystery solved...and this maybe something I can utilize in future experiments although as of right now I have no idea how. If we would have originally picked a rifS strepR colony for the fluctuation test, I wouldn't have realized any of this. Sometimes science is random.
We are currently trying to measure mutation rates to rifampicin resistance in Pseudomonas stutzeri. We already have encouraging results showing how a particular genotypic change increases mutation rates to streptomycin resistance and to goal is to calculate mutation rates an additional phenotype so we could begin to think that the genotypic change generally increases mutation rates. For other reasons, we wanted to measure the mutation rates to rifampicin resistance in a streptomycin resistant background strain.
Since P. stutzeri is competent for natural transformation, we transformed a rif and strep sensitive strain (rifS and strepS hereafter) using genomic DNA from a rif and strep resistant strain (rifR and strepR). No problem yet as we got plenty of strepR colonies back.
The next step is to use a fluctuation test to calculate mutation rates to rifR using cultures started from a single strepR colony. The basic idea of a fluctuation test is to grow many independent cultures starting from very low cell densities (typically ~1000 cells, in order to insure that there are no rifR colonies at the start), and then plate the entirety of these cultures under selective conditions once the cultures have grown to appreciable densities. If there are no rifR cells at the beginning of growth , you can use the distribution of rifR colonies that appear across the independent cultures in order to calculate mutation rates.
The picture below describes what you might expect from a typical fluctuation test.

When you plate out samples at time 0, there should be no rifR colonies (which I'm showing as blue circles if present). After growth, in this case 24 hours, rifR cells will have arisen by spontaneous mutation INDEPENDENTLY in each culture. As you can see from the picture, the expectation is that some cultures will not contain any rifR cells, some will have a few, and some will contain many rifR colonies. Those with many colonies are called "jackpots"and may appear as confluent lawns, hence the totality of blue in the picture. There is a distribution of rifR cells across independent cultures because mutations that lead to rifampicin resistance will occur at different points of the growth curve in each culture. A single mutation that arises during the first cell division after starting the experiment will lead to jackpots because these rifR cells have the chance to divide many times before plating. A rifR cell that arises during the last division before plating will only be represented by one colony because it hasn't had the time or resources to divide and proliferate. You can use this distribution to actually figure out mutation rates. Here is Stan Maloy's explanation of the fluctuation test.
Back to our P. stutzeri experiments...when we plated out the fluctuation test for our (what we thought) strepR rifS isolate, we got this result back from the time zero plate.

There was a lawn of rifR bacteria, which is statistically higher than zero (not really, but go with me on this). Even though the mutation rate to rifR is relatively high, something was fishy here because there should have been no colonies. We went back to the original plate of strepR transformants, and streaked additional isolates to both strep and rif plates. Even though the strain which we had originally transformed was rifS, I was surprised when roughly half of the strepR transformants were also rifR.
What could explain this result? The mutations that give rise to the strepR phenotype usually occur within a gene called rpsL. This gene codes for one of the many proteins that make up bacterial ribosomes, and indeed, the mechanism of action for streptomycin is inhibition of translation. The first thing I did was investigate the genomic context, what genes surround rpsL, within the one previously sequenced P. stutzeri genome. Here's what I found at the Pseudomonas genome database (which is extremely handy BTW).

All annotated genes in this section of the P. stutzeri genome are represented as red boxes. rpsL is the red box surrounded by a very dark line. The numbers on the bottom of the picture display the position of each gene in the P. stutzeri genome (rpsL is roughly at position 888,000 out of ~4,000,000). Here's where it gets interesting. Mutations that lead to rifampicin resistance typically occur in a gene called rpoB. This codes for a subunit of RNA polymerase, which makes sense (again) because the mechanism of action for rifampicin is to inhibit transcription. As I described above, we originally made the strepR strain by transforming a rifS strepS strain with genomic DNA from a rifR strepR strain. rpoB and rpsL are only about 5000 bp apart and I originally had no clue that rpoB and rpsL were genomic neighbors. Since, during natural transformation, genomic fragments sized 5Kb and above can be recombined into the recipient genome I'm guessing that a substantial fraction of the cells recombined both strepR and rifR mutations during this step from the donor genomic DNA. Since the exact positions of recombination are essentially random, cells that remained rifS after transformation must have recombined smaller fragments that only contained the strepR mutation. Here's a quick picture of what I think happened.

Rifampicin resistance is colored red, streptomycin resistance is colored blue. Cells that are rifR strepR are colored purple. While there were likely rifR strepS cells generated after transformation, these won't grow on the strepR plate so that's why there aren't red colonies. Mystery solved...and this maybe something I can utilize in future experiments although as of right now I have no idea how. If we would have originally picked a rifS strepR colony for the fluctuation test, I wouldn't have realized any of this. Sometimes science is random.
Friday, September 7, 2012
How I came to work with plant pathogens
It's getting to be grant season for me again, and one of the most important parts about writing grants is learning how to sell and justify your research story to a larger (somewhat less specialized) audience. I've also seen some stirrings on blogs and twitter about how to choose postdoc labs and research programs in order to set up an academic career. Although I think that the best answers to the postdoc question are much more dependent on both the individual and the lab, both of these topics inspired me to jot down some ramblings about how my own research program developed.
I've always been interested in understanding how microbial populations adapt to new environments and my graduate school career was spent studying evolutionary dynamics of the human pathogen Helicobacter pylori. Dr. Karen Guillemin had just started her lab at the University of Oregon and I'm most thankful that she was willing to take a chance on having an evolutionary biologist play around in her lab, which at the time was much more focused on understanding the molecular biology of bacterial pathogenesis. I was lucky to be co-advised by Dr. Patrick Phillips (a nematode guy), to fill in the blanks when it came to evolutionary biology, population genetics, and random Star Wars references. My grad school experiments centered around setting up a laboratory evolution system, modeled around the wonderful work of Rich Lenski, where I could test for the effects of genetic exchange on rates of adaptation. I'll talk about the ins and outs of those experiments in a future post, but at the end of graduate school I was left with a huge choice as to where to do my postdoc.At this time experimental evolution was exploding as a research field, and I wanted to continue studying bacterial adaptation using experimental evolution, but I also wanted to begin to study populations within hosts. I actually had a great interview with Rich Lenski in February 2006 in East Lansing where we talked about teaming up with Jeff Gordon at WashU to look at the effects of Rich's E. coli laboratory mutations during mouse infections (among other things including college basketball). After that interview, I was almost convinced that I was going to become a Michigan state Spartan.
There was a counter-voice in the back of my head, however, that maybe I should jump a little bit more out of my experimental comfort zone. I was feeling leery of working with mammalian pathogens for a variety of reasons: 1) I'm a fan of cute fuzzy animals like ferrets, and it would be very difficult for me to actually do research within hosts 2) Coming out of the Phillips lab I was well schooled in the importance of sample size for statistics. It's very expensive to perform large numbers of infections in anything with 4 legs and hair, and I was worried about getting high enough numbers of replicates to actually make sense of evolutionary trends. I knew I wanted to continue on in academia, and I had a serious internal discussion about how easy it would be to fund a lab that performed the experiments with mice that I was imagining. 3) I had a sneaky suspicion that genomics was about to explode (indeed, it already was in 2006), and I wanted to jump on the train. I certainly could have done this in Rich's lab but even at that point the field of the genomics of mammalian pathogens was getting crowded.
So what was my other option? I miraculously came across a random paper out of Jeff Dangl's lab, and had never heard of Dangl before this point (looking back...the only name I recognized on the paper was Dave Guttman from his work on recombination). I shot a quick email off to JD laying out my interests and and was pleasantly surprised that he was willing to fly me out for an interview in Chapel Hill. This interview was scheduled to be a couple of weeks after my interview in Michigan (in February). While can't say that the contrast in weather consciously affected my decision, it was 20 degrees and snowy in Michigan and 70 degrees and sunny in North Carolina during my visits. Dangl was well known in plant biology circles at that time for helping to work out the genetics of plant immune responses to pathogens, indeed, he was elected to the National Academy a couple of years later. Jeff was embarking on a pretty ambitious (as with seemingly all Dangl lab research) project to use "next-generation" sequencing technologies to illuminate genomic diversity in phylogenetically divergent strains of the plant pathogen Pseudomonas syringae. I knew that P. syringae was related to Pseudomonas fluorescens, which is a popular system for experimental evolution studies thanks to Paul Rainey and colleagues, and I figured that I might be able to piggy-back off of that system to set up my own P. syringae evolution experiments one day. This has actually proven to be spot on:) A postdoc at UNC would also throw me into the fire of bacterial genomics and force me to learn how to sequence and assemble strains, perform genomic level experiments, and PROGRAM! Perhaps most importantly, plants grow in dirt (which is dirt cheap) and no one really cares how or how many plants you euthanize during experiments so long as any transgenes stay contained. Working with plant pathogens would enable me to carry out an appropriate number of replicates to try and silence the subtle voice of Patrick Phillips that remains in my head today when I think about statistics and experimental design.
One other helpful piece of evidence that sealed the deal for me to join the Dangl lab was learning that mammalian and plant pathogens pretty much deploy the same sets of tools during pathogenesis. Evolutionary patterns from one system are very similar to the other and research findings are relevant across both, I wasn't really going to miss much studying plant pathogens instead of Shigella. For instance, the importance of type III secretion systems during infection was actually recognized at about the same time for both Yersinia pestis and P. syringae! The similarities have been laid out clearly in paper form a couple of times (here, here, here, here, here, among others) but the main differences between systems arise when immune responses in the hosts are compared. Even then, innate immunity is still a major shared component that contributes significantly to defense responses. As such I'm able to apply for grants across the main funding agencies (NSF, USDA, NIH), which theoretically makes make my life as a PI a little bit easier, although it hasn't yet:)
To be completely honest with you, I was torn after visiting both labs and took a couple of weeks to make my decision. What finally did it for me was flipping a coin, not once or twice, but until there was a run of the same side. UNC came up as the answer about 5 times in a row, and I found myself completely OK with that decision. Sometimes you just have to jump in and not look back.
I've always been interested in understanding how microbial populations adapt to new environments and my graduate school career was spent studying evolutionary dynamics of the human pathogen Helicobacter pylori. Dr. Karen Guillemin had just started her lab at the University of Oregon and I'm most thankful that she was willing to take a chance on having an evolutionary biologist play around in her lab, which at the time was much more focused on understanding the molecular biology of bacterial pathogenesis. I was lucky to be co-advised by Dr. Patrick Phillips (a nematode guy), to fill in the blanks when it came to evolutionary biology, population genetics, and random Star Wars references. My grad school experiments centered around setting up a laboratory evolution system, modeled around the wonderful work of Rich Lenski, where I could test for the effects of genetic exchange on rates of adaptation. I'll talk about the ins and outs of those experiments in a future post, but at the end of graduate school I was left with a huge choice as to where to do my postdoc.At this time experimental evolution was exploding as a research field, and I wanted to continue studying bacterial adaptation using experimental evolution, but I also wanted to begin to study populations within hosts. I actually had a great interview with Rich Lenski in February 2006 in East Lansing where we talked about teaming up with Jeff Gordon at WashU to look at the effects of Rich's E. coli laboratory mutations during mouse infections (among other things including college basketball). After that interview, I was almost convinced that I was going to become a Michigan state Spartan.
There was a counter-voice in the back of my head, however, that maybe I should jump a little bit more out of my experimental comfort zone. I was feeling leery of working with mammalian pathogens for a variety of reasons: 1) I'm a fan of cute fuzzy animals like ferrets, and it would be very difficult for me to actually do research within hosts 2) Coming out of the Phillips lab I was well schooled in the importance of sample size for statistics. It's very expensive to perform large numbers of infections in anything with 4 legs and hair, and I was worried about getting high enough numbers of replicates to actually make sense of evolutionary trends. I knew I wanted to continue on in academia, and I had a serious internal discussion about how easy it would be to fund a lab that performed the experiments with mice that I was imagining. 3) I had a sneaky suspicion that genomics was about to explode (indeed, it already was in 2006), and I wanted to jump on the train. I certainly could have done this in Rich's lab but even at that point the field of the genomics of mammalian pathogens was getting crowded.
So what was my other option? I miraculously came across a random paper out of Jeff Dangl's lab, and had never heard of Dangl before this point (looking back...the only name I recognized on the paper was Dave Guttman from his work on recombination). I shot a quick email off to JD laying out my interests and and was pleasantly surprised that he was willing to fly me out for an interview in Chapel Hill. This interview was scheduled to be a couple of weeks after my interview in Michigan (in February). While can't say that the contrast in weather consciously affected my decision, it was 20 degrees and snowy in Michigan and 70 degrees and sunny in North Carolina during my visits. Dangl was well known in plant biology circles at that time for helping to work out the genetics of plant immune responses to pathogens, indeed, he was elected to the National Academy a couple of years later. Jeff was embarking on a pretty ambitious (as with seemingly all Dangl lab research) project to use "next-generation" sequencing technologies to illuminate genomic diversity in phylogenetically divergent strains of the plant pathogen Pseudomonas syringae. I knew that P. syringae was related to Pseudomonas fluorescens, which is a popular system for experimental evolution studies thanks to Paul Rainey and colleagues, and I figured that I might be able to piggy-back off of that system to set up my own P. syringae evolution experiments one day. This has actually proven to be spot on:) A postdoc at UNC would also throw me into the fire of bacterial genomics and force me to learn how to sequence and assemble strains, perform genomic level experiments, and PROGRAM! Perhaps most importantly, plants grow in dirt (which is dirt cheap) and no one really cares how or how many plants you euthanize during experiments so long as any transgenes stay contained. Working with plant pathogens would enable me to carry out an appropriate number of replicates to try and silence the subtle voice of Patrick Phillips that remains in my head today when I think about statistics and experimental design.
One other helpful piece of evidence that sealed the deal for me to join the Dangl lab was learning that mammalian and plant pathogens pretty much deploy the same sets of tools during pathogenesis. Evolutionary patterns from one system are very similar to the other and research findings are relevant across both, I wasn't really going to miss much studying plant pathogens instead of Shigella. For instance, the importance of type III secretion systems during infection was actually recognized at about the same time for both Yersinia pestis and P. syringae! The similarities have been laid out clearly in paper form a couple of times (here, here, here, here, here, among others) but the main differences between systems arise when immune responses in the hosts are compared. Even then, innate immunity is still a major shared component that contributes significantly to defense responses. As such I'm able to apply for grants across the main funding agencies (NSF, USDA, NIH), which theoretically makes make my life as a PI a little bit easier, although it hasn't yet:)
To be completely honest with you, I was torn after visiting both labs and took a couple of weeks to make my decision. What finally did it for me was flipping a coin, not once or twice, but until there was a run of the same side. UNC came up as the answer about 5 times in a row, and I found myself completely OK with that decision. Sometimes you just have to jump in and not look back.
Monday, August 27, 2012
On the ESA letter to NSF
As many of you may know (and many of you may not), there's a draft letter circulating around the ecology/evolution community concerning reforms made to the NSF grant proposal system. Proflikesubstance has been much more on top of this, so for the relevant background please start HERE and work your way back.
First off, I signed the letter. While many (including some of the people I respect most in science) say that the letter doesn't offer solutions, a sentiment I agree with, I do see it as the next step in the debate. This letter gives a formal voice to the "opposition", rather than a bunch of us ragging and complaining to colleagues and the ethos. Some will say it's whiney, but debate is about voices on both sides of issues. Again, I wholly agree that the NSF didn't have a choice since the previous system was unsustainable. There is not as much money going around as we'd like, and there are many more scientific mouths to fill now than before. I think eventually we will reach a point where all sides are content, not happy but content, but it's going to take a while and bits and pieces of solutions from all sides to get to that point. I also think as scientists we are going to need to chip in a little bit more to make the system work, see below, without necessarily getting anything back aside from helping to make the community of peers a better place.
As a young PI, I have a hell of a lot of skin in this game. While I don't have a complete solution to the problem, there are a couple of ways that the system seriously complicates life as an assistant professor and I think these issues could be addressed better. The two main complications in my own experience are the PI limit on submissions and the lack of critique from the combination of pre-proposals plus only one submission round a year. This is maybe my naivety in the situation, but I don't understand why there is a limit. Can somebody in the comments please tell me what this is trying to address? All I can speak from is brief experience, but I've had more than a couple of situations in the last year where those who could act as collaborators with me (and will act as collaborators anyway) have worried about putting their names on my grants because of the limit. These folks are going to help me with the project regardless. When I submit grants on these collaborative projects, I worry that it seems as though my submission and accompanying letters of support don't fully portray the potential success for these projects and don't accurately portray the skill sets behind the submissions. I certainly don't have extensive experience with every single scientific technique or analysis, but when I include experiments utilizing these techniques into my proposals I can't help but think that there is an inherent psychological disadvantage leaving collaborator's names off. This may be a problem only within my own head, but PI's are having to count the number of submissions they are on and worry about prioritizing the limited submission opportunities and this definitely trickles through.
The biggest issue in my own experience is the lack of feedback/critique that young PI's get on proposals. Pre-tenure, we as assistant professors have a set amount of time to make our case that we belong as PIs. This means becoming the best grant writer we can possibly be as quickly as possible, and this only happens with practice. Currently, there is one submission per year to IOS and DEB. From what Proflikesubstance says in the link above it seems like MCB is going to follow suit. I started my lab roughly two years ago, and have roughly 3 1/2 years left until my tenure package is due. Therefore, for the NSF, I've got roughly 3 more pre-proposal submission opportunities per panel. Pre-proposals are not full grants, and this severely curtails the opportunity to go into the experimental details. There is simply no way that I will be able to get the feedback that I would have under the previous system just through the pre-proposals. If you get selected to submit a full proposal, you will still receive half of the potential critique that you will have gotten previously given one funding opportunity per year instead of 2. One of the greatest challenges to starting a lab is learning how to write and sell your grants to a broad audience and developing your ideas. Simply as a product of time on the job, many of your grant submissions and research ideas haven't been vetted as much as those of more established PIs. Every time I've written grants my first two years on the job, they have been made up from scratch. Only now do I have enough submissions under my belt where I can start to distill ideas and pull language from the grants that work while dropping or refining sentences that don't. I have great mentors here at UA and many more friends at other institutions that have helped, but there is something special about anonymous review that makes these critiques extra valuable for developing grants and research programs (harsh as they may be). Critique from failed NSF research submissions turns into better NSF grants later, but it also feeds NIH/USDA/etc... grants later. Limiting the amount of critique I get from the full submissions by anonymous reviewers significantly affects my grant submissions to all funding agencies. Others may counter with the idea young PIs have the unique ability to submit CAREER proposals and this is certainly true. However, these proposals are skewed much more to teaching and outreach than normal grants (further limiting space for vetting experiments and changing experimental focus) and there's a total limit on the number of CAREER proposals per PI over their eligibility. I was reluctant to submit a CAREER this year (I strongly thought about it just to get reviews back) because I haven't had much time to figure out the best ways to incorporate my research into teaching and outreach...while also thinking that I would have a better chance preliminary data-wise if I waited til my third year. In the end, I didn't think the numbers worked out in my favor given this cost-benefit analysis for CAREER submission this summer but with this decision I lose another chance at critique as a young PI.
The ESA letter is a bit weak on solutions, yes. Barring a good reason for the PI limit I don't see why this can't be raised or eliminated without affecting the system much. Modern science is based on collaboration and we should celebrate this. If there is a "too many submissions from PIs" problem, can't we come to some agreement on how not to exploit the system? Maybe I'm an idealist... As for the critique problem, maybe the solution is having an NSF workshop for young PIs every year that acts essentially as a panel. Young PIs and established PIs could volunteer to act on this workshop panel, where the focus would be on anonymous critique of proposals from young PIs. I've noticed that many young PIs have been encouraged to act on NSF panels, and this provides yet another opportunity (I've volunteered for numerous separate panels thus far, but each had been previously filled). This panel could be timed to provide relevant feedback to pre and full-proposals for the next round of submissions. While no grant money would be handed out, this would provide more opportunities for anonymous critique while also providing a deadline to shoot for and insights into the granting process. It might even be worthwhile if we could get our institutions to buy into these panels as a form of service?
Anyway, my two cents. I have a great deal of optimism that we can figure something out that satisfies all parties, while agreeing that the old system was unsustainable avoiding just reverting to the comfortable past. The NSF is different than many other negotiations (that's a crappy word for it but two sides disagreeing and coming to the best solution) because program officers are scientists and understand the perils and worries of life as researchers. I signed the letter because it solidifies an opposing voice even if it doesn't go anywhere, but it's a start.
First off, I signed the letter. While many (including some of the people I respect most in science) say that the letter doesn't offer solutions, a sentiment I agree with, I do see it as the next step in the debate. This letter gives a formal voice to the "opposition", rather than a bunch of us ragging and complaining to colleagues and the ethos. Some will say it's whiney, but debate is about voices on both sides of issues. Again, I wholly agree that the NSF didn't have a choice since the previous system was unsustainable. There is not as much money going around as we'd like, and there are many more scientific mouths to fill now than before. I think eventually we will reach a point where all sides are content, not happy but content, but it's going to take a while and bits and pieces of solutions from all sides to get to that point. I also think as scientists we are going to need to chip in a little bit more to make the system work, see below, without necessarily getting anything back aside from helping to make the community of peers a better place.
As a young PI, I have a hell of a lot of skin in this game. While I don't have a complete solution to the problem, there are a couple of ways that the system seriously complicates life as an assistant professor and I think these issues could be addressed better. The two main complications in my own experience are the PI limit on submissions and the lack of critique from the combination of pre-proposals plus only one submission round a year. This is maybe my naivety in the situation, but I don't understand why there is a limit. Can somebody in the comments please tell me what this is trying to address? All I can speak from is brief experience, but I've had more than a couple of situations in the last year where those who could act as collaborators with me (and will act as collaborators anyway) have worried about putting their names on my grants because of the limit. These folks are going to help me with the project regardless. When I submit grants on these collaborative projects, I worry that it seems as though my submission and accompanying letters of support don't fully portray the potential success for these projects and don't accurately portray the skill sets behind the submissions. I certainly don't have extensive experience with every single scientific technique or analysis, but when I include experiments utilizing these techniques into my proposals I can't help but think that there is an inherent psychological disadvantage leaving collaborator's names off. This may be a problem only within my own head, but PI's are having to count the number of submissions they are on and worry about prioritizing the limited submission opportunities and this definitely trickles through.
The biggest issue in my own experience is the lack of feedback/critique that young PI's get on proposals. Pre-tenure, we as assistant professors have a set amount of time to make our case that we belong as PIs. This means becoming the best grant writer we can possibly be as quickly as possible, and this only happens with practice. Currently, there is one submission per year to IOS and DEB. From what Proflikesubstance says in the link above it seems like MCB is going to follow suit. I started my lab roughly two years ago, and have roughly 3 1/2 years left until my tenure package is due. Therefore, for the NSF, I've got roughly 3 more pre-proposal submission opportunities per panel. Pre-proposals are not full grants, and this severely curtails the opportunity to go into the experimental details. There is simply no way that I will be able to get the feedback that I would have under the previous system just through the pre-proposals. If you get selected to submit a full proposal, you will still receive half of the potential critique that you will have gotten previously given one funding opportunity per year instead of 2. One of the greatest challenges to starting a lab is learning how to write and sell your grants to a broad audience and developing your ideas. Simply as a product of time on the job, many of your grant submissions and research ideas haven't been vetted as much as those of more established PIs. Every time I've written grants my first two years on the job, they have been made up from scratch. Only now do I have enough submissions under my belt where I can start to distill ideas and pull language from the grants that work while dropping or refining sentences that don't. I have great mentors here at UA and many more friends at other institutions that have helped, but there is something special about anonymous review that makes these critiques extra valuable for developing grants and research programs (harsh as they may be). Critique from failed NSF research submissions turns into better NSF grants later, but it also feeds NIH/USDA/etc... grants later. Limiting the amount of critique I get from the full submissions by anonymous reviewers significantly affects my grant submissions to all funding agencies. Others may counter with the idea young PIs have the unique ability to submit CAREER proposals and this is certainly true. However, these proposals are skewed much more to teaching and outreach than normal grants (further limiting space for vetting experiments and changing experimental focus) and there's a total limit on the number of CAREER proposals per PI over their eligibility. I was reluctant to submit a CAREER this year (I strongly thought about it just to get reviews back) because I haven't had much time to figure out the best ways to incorporate my research into teaching and outreach...while also thinking that I would have a better chance preliminary data-wise if I waited til my third year. In the end, I didn't think the numbers worked out in my favor given this cost-benefit analysis for CAREER submission this summer but with this decision I lose another chance at critique as a young PI.
The ESA letter is a bit weak on solutions, yes. Barring a good reason for the PI limit I don't see why this can't be raised or eliminated without affecting the system much. Modern science is based on collaboration and we should celebrate this. If there is a "too many submissions from PIs" problem, can't we come to some agreement on how not to exploit the system? Maybe I'm an idealist... As for the critique problem, maybe the solution is having an NSF workshop for young PIs every year that acts essentially as a panel. Young PIs and established PIs could volunteer to act on this workshop panel, where the focus would be on anonymous critique of proposals from young PIs. I've noticed that many young PIs have been encouraged to act on NSF panels, and this provides yet another opportunity (I've volunteered for numerous separate panels thus far, but each had been previously filled). This panel could be timed to provide relevant feedback to pre and full-proposals for the next round of submissions. While no grant money would be handed out, this would provide more opportunities for anonymous critique while also providing a deadline to shoot for and insights into the granting process. It might even be worthwhile if we could get our institutions to buy into these panels as a form of service?
Anyway, my two cents. I have a great deal of optimism that we can figure something out that satisfies all parties, while agreeing that the old system was unsustainable avoiding just reverting to the comfortable past. The NSF is different than many other negotiations (that's a crappy word for it but two sides disagreeing and coming to the best solution) because program officers are scientists and understand the perils and worries of life as researchers. I signed the letter because it solidifies an opposing voice even if it doesn't go anywhere, but it's a start.
Wednesday, August 22, 2012
Follow the biology (pt. 3), slight end of summer update
I'd like to apologize first about how sparse my postings have been, but, starting this blog just happened to coincide with the summer conference season as well as the start of the semester here at U of A.
I thought I'd jump back into this by giving a very brief update on an open science project that I started to talk about a few weeks ago (parts. 1 here and 2 here). Over the summer I had an undergrad working on the project in order to isolate transposon mutants that don't display the brown phenotype. After screening ~1000 independent colonies, I'm sad to say that every single plate looked exactly like this:

There were always a couple of empty wells where the colonies didn't grow but, eventually, every well turned brown. Astute readers may point out that well A4 doesn't look so brown (hence why I have this picture lying around). However, even this well turned brown after an additional day of growth....and every single colony isolated from that well (~20 or so) turned brown so the late browning wasn't just contamination. Of course, it's possible that whatever pathway leads to the brown phenotype is essential for cellular growth so that mutations would never be identified from a transposon screen. 1000 colonies isn't enough to make a case either way, but it's enough to begin thinking about the next step.
Now that the summer is over, the talented undergrad working on that project has moved on to grad school in another department so I hesitate to devote much more time to the transposon hunt. While it's possible that I could eventually isolate a non-brown colony, there are better uses of my time right now. It was worth a shot but this is a point in my own brain where I cut bait and try a different strategy (especially when the undergrad has moved on to another department). So now I'm going to isolate some genomic DNA from this mutant and sequence the genome in order to identify any potential genotypic differences. We'll see how that goes.
I thought I'd jump back into this by giving a very brief update on an open science project that I started to talk about a few weeks ago (parts. 1 here and 2 here). Over the summer I had an undergrad working on the project in order to isolate transposon mutants that don't display the brown phenotype. After screening ~1000 independent colonies, I'm sad to say that every single plate looked exactly like this:

There were always a couple of empty wells where the colonies didn't grow but, eventually, every well turned brown. Astute readers may point out that well A4 doesn't look so brown (hence why I have this picture lying around). However, even this well turned brown after an additional day of growth....and every single colony isolated from that well (~20 or so) turned brown so the late browning wasn't just contamination. Of course, it's possible that whatever pathway leads to the brown phenotype is essential for cellular growth so that mutations would never be identified from a transposon screen. 1000 colonies isn't enough to make a case either way, but it's enough to begin thinking about the next step.
Now that the summer is over, the talented undergrad working on that project has moved on to grad school in another department so I hesitate to devote much more time to the transposon hunt. While it's possible that I could eventually isolate a non-brown colony, there are better uses of my time right now. It was worth a shot but this is a point in my own brain where I cut bait and try a different strategy (especially when the undergrad has moved on to another department). So now I'm going to isolate some genomic DNA from this mutant and sequence the genome in order to identify any potential genotypic differences. We'll see how that goes.
Tuesday, July 10, 2012
Follow the biology (pt. 2)...Why not sequence that thing?
I spent a good part of my time as a postdoc figuring out how to sequence microbial genomes cheaply, and learning how to deal with the flood of data that ensued. After my first post on this project, I was asked a very good and relevant question by Olin Silander...specifically, why not just spend a couple of hundred dollars and sequence the interesting brown mutant rather than go through transposon mutagenesis? I'm guessing that if you're reading this and are somewhat up to date on modern microbiology, you probably have the same question. Here's a couple of reasons that I came up with with varying levels of importance:
1) I'm a sucker for old school genetic screens. To be honest, it's fun coming into the lab and checking whether cultures are brown or yellow. There's so much about being a PI that can stress you out, and that feeling of "I got one!" is one little simple pleasure that I love about research. Agar plates, media, and bacterial conjugations are cheap...and I've got a very talented recently post-grad summer student that's helping me out with this experiment to ease the time investment so it's really not a big waste of anything.
2) It's possible that sequencing and transposon mutagenesis will yield different results. Whereas sequencing will likely tell me exactly what the mutation is, it's possible (maybe likely given that my gut is telling me that the brown color is a product of overproduction or disruption of negative regulation) that the exact mutation yielding the brown phenotype is in some regulatory region/protein. However, I'll admit that it is equally as likely at this moment that the brown phenotype is due to disruption of an enzymatic pathway and that what I'm seeing is the buildup of an intermediate product that is getting oxidized. IF the mutation is regulatory though, the transposon screen will likely yield disruptions in the pathways producing the brown product and that act downstream of the initial mutation. With only the sequence of a regulatory mutation I could be left guessing what pathway was being affected and what the next step is.
3) Transposon disruptions give me knockout mutants, and I can directly verify cause and effect of the transposon disruption on knocking out the brown coloration by transforming these loci back into the original brown mutant. If this ever is publishable, I need to be able to demonstrate genetic causation of a phenotype and natural transformation of the brown mutant with an antibiotic resistance locus present within a transposon is slightly easier than the molecular biology gymnastics necessary to recreate the original brown mutant in a wild type background. Additionally, more phenotypes (in this case disruption of the brown color) are never bad things.
4) This one is a little inside baseball...but I'm teaching upper level microbial genetics next spring with a lab (about 80 students in the lab). As part of this lab, I want to have the students perform transposon mutagenesis and go over basic ideas like screens, selections, phenotypes. This little project gives me the opportunity to fine tune this procedure in P. stutzeri using an easily scoreable visual readout and provides a good base for figuring out what experiments I can have the students perform next April.
5) I'm going to sequence it:) However, given how much of an overkill it is to sequence bacterial genomes using Illumina's HiSeq technology, I'm waiting to collect 23 other bacterial genomes to sequence along this one (I've got about 17 right now) in a single lane. I'll be blogging about those results too, so don't worry.
1) I'm a sucker for old school genetic screens. To be honest, it's fun coming into the lab and checking whether cultures are brown or yellow. There's so much about being a PI that can stress you out, and that feeling of "I got one!" is one little simple pleasure that I love about research. Agar plates, media, and bacterial conjugations are cheap...and I've got a very talented recently post-grad summer student that's helping me out with this experiment to ease the time investment so it's really not a big waste of anything.
2) It's possible that sequencing and transposon mutagenesis will yield different results. Whereas sequencing will likely tell me exactly what the mutation is, it's possible (maybe likely given that my gut is telling me that the brown color is a product of overproduction or disruption of negative regulation) that the exact mutation yielding the brown phenotype is in some regulatory region/protein. However, I'll admit that it is equally as likely at this moment that the brown phenotype is due to disruption of an enzymatic pathway and that what I'm seeing is the buildup of an intermediate product that is getting oxidized. IF the mutation is regulatory though, the transposon screen will likely yield disruptions in the pathways producing the brown product and that act downstream of the initial mutation. With only the sequence of a regulatory mutation I could be left guessing what pathway was being affected and what the next step is.
3) Transposon disruptions give me knockout mutants, and I can directly verify cause and effect of the transposon disruption on knocking out the brown coloration by transforming these loci back into the original brown mutant. If this ever is publishable, I need to be able to demonstrate genetic causation of a phenotype and natural transformation of the brown mutant with an antibiotic resistance locus present within a transposon is slightly easier than the molecular biology gymnastics necessary to recreate the original brown mutant in a wild type background. Additionally, more phenotypes (in this case disruption of the brown color) are never bad things.
4) This one is a little inside baseball...but I'm teaching upper level microbial genetics next spring with a lab (about 80 students in the lab). As part of this lab, I want to have the students perform transposon mutagenesis and go over basic ideas like screens, selections, phenotypes. This little project gives me the opportunity to fine tune this procedure in P. stutzeri using an easily scoreable visual readout and provides a good base for figuring out what experiments I can have the students perform next April.
5) I'm going to sequence it:) However, given how much of an overkill it is to sequence bacterial genomes using Illumina's HiSeq technology, I'm waiting to collect 23 other bacterial genomes to sequence along this one (I've got about 17 right now) in a single lane. I'll be blogging about those results too, so don't worry.
Friday, July 6, 2012
Follow the biology (pt I)
Being a PI can be stressful for a variety of reasons, but I love my job as a researcher. There is no greater thrill for a curious mind than asking questions, designing experiments, and figuring out how nature works. Partly inspired by Rosie Redfield, one of my motivations for writing this blog is to relay the fun/not so fun moments of everyday science. In the spirit of openness I'm periodically going to be writing about a little side project that I've got going on in my lab. I actually have no clue where this will go, and at the moment there is absolutely no hypothesis, but I'm curious to see how this turns out because we've found something phenotypically interesting (to us at least). Science is about asking questions after all, and following the biology (currently reading an ASM book dedicated to John Roth, and "Follow the biology" is advice John liked to give to folks in his lab).

So, to begin, one of the bacteria that my lab works on is Pseudomonas stutzeri. The strain that we've been focusing much of our attention on usually makes vibrant yellow colonies during growth on agar plates, and this yellow tinge also comes through during growth in liquid culture (see test tube on left below). From my other work on Pseudomonads, I'm guessing that these colors might be due to iron scavenging molecules called siderophores, but I haven't had time to read a lot about the genetics of color in P. stutzeri, or even find out if tere are things to read. One day my technician picked a single colony from this workhorse strain and grew up an overnight culture. There was nothing particularly special about this colony or the overnight culture, but the next day this overnight culture was placed in the fridge to save for later. My technician later took the culture out of the fridge and, very surprisingly, noticed that the culture was now dark brown instead of the yellow tinge. We isolated single colonies from this brown culture and, sure enough, they continue to turn liquid overnight cultures brown. When colonies from this line are grown on plates, you can tell that the brown color is due to something extracellular to the colonies and which diffuses throughout the agar. Somehow, we were lucky (?) enough to randomly pick the very colony that possessed a mutation leading to an interesting phenotype (the odds are hugely against that). It's also not simply a contaminant...ruled that out already.

Culture on the left is "wild type" P. stutzeri, on the right is our weird, brown culture isolate
What's the first step of figuring out what this phentoype is (well, the first step is actually blogging about it and seeing if anyone has ever witnessed this before...if you have please let me know). If I were a biochemist I might try to isolate the chemical that's turning the culture brown, do some fancy analysis, and figure out what the composition is. I'm not a biochemist, I'm a geneticist. We like to figure out the underlying genes involved by breaking things and cleaning up the mess. To do this I am going to be using a Tn5 transposon. Transposons are pieces of DNA found widely in nature and can be thought about as chromosomal parasites. Tn5 is a naturally occurring transposon that has been "domesticated" for use in genetics labs. Once in a bacterial cell, it will randomly "hop" or "transpose" into the chromosome of the host. A good metaphor for this is that it cuts itself out of whatever previous piece of DNA it is present in, and pastes itself into the bacterial chromosome. The Tn5 transposon will hop only once into a (for the most part) random section of the new chromosome. Incorporation of the Tn5 will usually disrupt the function of whatever gene it incorporates into. We are going to hop a Tn5 transposon into the brown P. stutzeri isolates chromosome, with the hopes that this Tn5 will disrupt whatever genes are making the culture brown.

Overview of our Tn5 strategy
The particular strain of P. stutzeri that we are working with is competent for natural transformation in the lab, which basically means that it has the ability to suck up pieces of DNA from the extracellular environment and recombine them into it's own genome so long as there is some sequence similarity. Once we isolate a strain containing a transposon that eliminates the brown phenotype, we want to be sure that the transposon is causative for this phenotype rather than some other unknown mutation. To confirm this, we will isolate genomic DNA from the transposon mutant and transform the original brown strain with genomic DNA containing the transposon. If this Tn5 disruption is causative, we will be able to select for transformants that are no longer brown. Then, all thats left is to figure out what genes the transposon disrupted. We can do this using a few genetic tricks that I'll talk about later, but currently we are still trying to isolate non-brown isolates.
Wednesday, July 4, 2012
Selection-driven Gene Loss in Bacteria
Bacterial genomes differ dramatically in size: from 140Kb to 13Mb (those numbers might be off now...please let me know if something has broken the record. Yes, I know the lower estimate can change based on semantics, but there are a bunch in that range). Although we have some clues as to how selection acts (or fails to act) on genome size, outside of intracellular parasites it's a bit of a mystery how selection shapes total genomic content. Perhaps the most interesting case out there involves genome streamlining in marine bacteria, which has been attributed to selection but which remains a just so story to this point.



One easy explanation is that "extra" DNA is costly in and of itself because it takes things like carbon, phosphorous, and nitrogen to physically make DNA. While probably true in the strictest sense, as far as I know there has not been a very clear test of the actual selective forces that act at this level. I would guess, especially given what's come out of the arsenic life debacle, that bacterial cells can survive just fine the way they are with low levels of phosphorous et al. I'm not sure how many environments are limiting enough for these elements to have direct selective effects on genome size (although see situations like this, this, and this). Other recent research points to the "cost" of extra DNA residing in the production of RNA and proteins. In this experiment, proteins and ribonucleotides are not inherently costly (*under the environments tested), but production of unnecessary proteins likely takes away cellular machinery that could otherwise be put to better use. There are only so many ribosomes in a cell to carry out translation. If these are occupied by unnecessary transcripts, they can't be used to produce more essential proteins. Interestingly, these costs may change based on previous environment. There are also some additional other hypotheses for genome size evolution that I may touch on in the future, but for now I would like to give a brief overview and thoughts about a paper relevant to this question that came out last week in PLoS Genetics from Dan Andersson's group.

Schematic of how they isolated deletion mutants
This manuscript is basically laid out in three related, but independent parts. The first consists of measuring the rate of deletions throughout the Salmonella enterica var typhimurium LT2 chromosome (see figure above). They first hopped a transposon containing three phenotypic markers into random areas of the chromosome. The markers contribute 1) resistance to chloramphenicol 2) cleavage of B-galactosidase leading to blue colonies during growth on X-gal 3) sensitivity to chlorate. They can measure rates of deletion using this transposon because of a cool genetic trick: the moaA marker renders the cells sensitive to chlorate, so they can select on resistance to chlorate in order to identify when the transposon might have been deleted from the chromosome. Once they get these chlorate resistance mutants, they look for white colonies and those that are sensitive to chloramphenicol in order to eliminate cells that have only deleted small portions of the transposon or have inactivated moaA through mutation.

The "deletometer" transposon
They use this "deletometer" to measure rates of mutation in 11 chromosomal regions and find that this rate varies by 2 orders of magnitude. These deletions range up to 10's of thousands of bp in size. Kind of a sidenotes to the total story here, but they do find evidence for the existence of a RecA independent deletion mechanism in S. enterica by studying genomic context of the deletions (RecA needs 25bp of sequence similarity to recombine pieces of DNA, but they find evidence that there is much less similarity bordering many of their sampled deletions, and in some cases none). They also find that, as shown for other bacteria like Bordetalla, that the replication terminus seems to be a hotspot for deletions (higher rates at terminus). The one question in my mind that remains from this portion of the paper is how they control for genomic context. It seems like regions of the chromosome that have more redundant sequence should have higher deletion rates, but maybe I haven't thought through this enough.

Fitness effects of deletions across enviroments and assays
Next they use a subset isolates to test for fitness effects of the deletions (and therefore address the question of cost of extra DNA). By measuring growth of strains in two environments (rich and minimal media) they show that some of the deletions actually increase growth rate in 15 of 55 cases. They further reinforce that deletions can be beneficial using growth assays where two strains are directly competed against one another. Importantly, they find no relationship between the size of deletion and the fitness effects (strike for the DNA is costly in and of itself camp).
The third part of the manuscript consists of a 1000 generation passage experiment in rich media. Over the course of laboratory passage and adaptation, there are a suite of deletions that reach high frequencies and are therefore likely adaptive during lab passage and in the right genomic context. They recreate these deletions in an unadapted chromosome and are able to show that 2 out of 6 do increase bacterial fitness in the lab (the absence of effects for the remaining four they chalk up to epistasis...they are beneficial only in the presence of other mutations that are not present in the unadapted ancestral strain).
The overall, memorable, take home message from this paper is that random deletions can be beneficial under some circumstances. Although this has been previously seen, this paper extends the result. They don't test the mechanistic underpinnings of these selective effects, which is what I hoped might be in the paper given the title. Although the authors don't talk about this too much, there a lot of deletions that are detrimental under each condition. The data seems to indicate that specific regions of the chromosome are more costly than others (beneficial fitness effects are only found in a subset of chromosomal positions), and I'm curious whether there is some unifying theme to these regions. Maybe they contain highly expressed but unnecessary, and therefore wasteful, genes. I'm curious why there are differential fitness effects for mutations that affect the same region...seems like it would be straightforward to figure out what differs amongst these different deletions within the same region as a way to get at the cost of DNA question. There is some additional novelty in showing that deletion rate varies over the chromosome, that there seems to be a RecA independent deletion mechanism in S. enterica, and that there is a deletion hotspot in the terminus.
Here's the citation:
Koskiniemi S, Sun S, Berg OS, Andersson DI. 2012 "Selection-Driven Gene Loss in Bacteria". PLoS Genetics.
Here's the citation:
Koskiniemi S, Sun S, Berg OS, Andersson DI. 2012 "Selection-Driven Gene Loss in Bacteria". PLoS Genetics.
Thursday, June 28, 2012
What is a bacterial population?
One of the most important parameters within population genetics is effective population size (Ne). Ne determines how strong genetic drift is within populations, and therefore how weakly selection sorts among genotypes. I'm not going to go through a bunch of examples of why it's such an important parameter (maybe later), suffice to say that Ne calculations can be used to explain results such as the origin of genome complexity as well as establishing divergence times between bacteria (such as here). Slight digression: I absolutely hate when divergence times are put on bacterial lineages. Unlike many eukaryotic taxa, bacterial populations don't leave easily dateable fossils so divergence times are completely based on population genetic estimates.
Which brings me back to Ne. Effective population size is just that, a measurement of population size. The problem I have with using this parameter in microbial populations is that I (and I suspect everyone else) don't have a clue as to what constitutes a microbial population. Huge numbers always get thrown around about how many bacterial cells exist in nature (10 times as many bacterial cells as human cells in the body!). Are all these cells one population? Are all E. coli cells within your colon one population? Any estimation of Ne is just a guess because it's hard to define what a population is and population sizes likely vary over many orders of magnitude. I can be wrong about that, but that's why I'm putting my thoughts out there.
The best place to start tackling this question is in obligate parasites/symbionts. We have good evidence that vertically inherited symbionts such as Buchnera aphidicola have small Ne values because genetic drift has transformed their genomes. Likewise, it might be possible to define what populations are for certain obligate parasites (such as my good friend Helicobacter pylori, which can really only survive inside the human stomach) that are only found in closed environments. Even in cases such as H. pylori, transmission across hosts from stomach to stomach starts to blur the lines between populations. As far as I can tell the situation gets much more difficult to model as you step from generalist pathogens/symbionts to environmental bacteria to spore formers (or persisters) that can survive in environments without growing. What about population subdivision such as in biofilms? What about dramatic lifestyle changes and population bottlenecks such as in Vibrio fischeri. V. fischeri can be found living freely as saprophytes in the open ocean (see link within here), but all it takes is one single bacterium to infect a juvenile squid and be amplified by the billions. Bottlenecking from millions of cells to 1 dramatically altars Ne since calculations of this parameter place extra weight on low numbers.
Unlike what is possible with more visible megafauna, we can't simply go out and perform ecological catch and release studies to estimate population sizes. The best way to think about microbial populations may be, somewhat abstractly, in population genetic terms. If a new mutation arises, what other genotypes are going to affect its rates of fixation or persistance? If genetic variants arise within competing evolutionary backgrounds and directly affect each others frequencies, I take that as good evidence that those competing backgrounds are in the same population. Likewise, if the rates of migration/transmission between hosts or subpopulations significantly affects selection then these subpopulations could be considered parts of a larger whole.
 As, hopefully, illustrated in the figure above, I think of microbial populations as a continuum. On the left side is one closed population where three genotypes (red, blue, green) all directly compete for resources and affect the frequencies of one another. On the right side these three genotypes are completely separate from one another and don't directly interact. My guess is that most microbial populations lie somewhere in the middle, where there is some subdivision but migration and transmission between subpopulations alters selection and genetic drift.
As, hopefully, illustrated in the figure above, I think of microbial populations as a continuum. On the left side is one closed population where three genotypes (red, blue, green) all directly compete for resources and affect the frequencies of one another. On the right side these three genotypes are completely separate from one another and don't directly interact. My guess is that most microbial populations lie somewhere in the middle, where there is some subdivision but migration and transmission between subpopulations alters selection and genetic drift.
So how do we begin to measure microbial populations? I don't think it's been done yet, (please feel free to correct me!), but the rise of metagenomics at least makes the necessary experiments possible. With these technologies we can measure genotypes over time (all genotypes, not just a culturable subsample). We can measure how genotypes affect frequencies of other genotypes, and interactions could be evidence of existing within the same population. We could measure parameters such as strength of genetic drift and selection over time and extrapolate back to get Ne. Of course we then get into the issue of species level interactions, but I'll definitely save that for another time.
Thoughts?
Which brings me back to Ne. Effective population size is just that, a measurement of population size. The problem I have with using this parameter in microbial populations is that I (and I suspect everyone else) don't have a clue as to what constitutes a microbial population. Huge numbers always get thrown around about how many bacterial cells exist in nature (10 times as many bacterial cells as human cells in the body!). Are all these cells one population? Are all E. coli cells within your colon one population? Any estimation of Ne is just a guess because it's hard to define what a population is and population sizes likely vary over many orders of magnitude. I can be wrong about that, but that's why I'm putting my thoughts out there.
The best place to start tackling this question is in obligate parasites/symbionts. We have good evidence that vertically inherited symbionts such as Buchnera aphidicola have small Ne values because genetic drift has transformed their genomes. Likewise, it might be possible to define what populations are for certain obligate parasites (such as my good friend Helicobacter pylori, which can really only survive inside the human stomach) that are only found in closed environments. Even in cases such as H. pylori, transmission across hosts from stomach to stomach starts to blur the lines between populations. As far as I can tell the situation gets much more difficult to model as you step from generalist pathogens/symbionts to environmental bacteria to spore formers (or persisters) that can survive in environments without growing. What about population subdivision such as in biofilms? What about dramatic lifestyle changes and population bottlenecks such as in Vibrio fischeri. V. fischeri can be found living freely as saprophytes in the open ocean (see link within here), but all it takes is one single bacterium to infect a juvenile squid and be amplified by the billions. Bottlenecking from millions of cells to 1 dramatically altars Ne since calculations of this parameter place extra weight on low numbers.
Unlike what is possible with more visible megafauna, we can't simply go out and perform ecological catch and release studies to estimate population sizes. The best way to think about microbial populations may be, somewhat abstractly, in population genetic terms. If a new mutation arises, what other genotypes are going to affect its rates of fixation or persistance? If genetic variants arise within competing evolutionary backgrounds and directly affect each others frequencies, I take that as good evidence that those competing backgrounds are in the same population. Likewise, if the rates of migration/transmission between hosts or subpopulations significantly affects selection then these subpopulations could be considered parts of a larger whole.
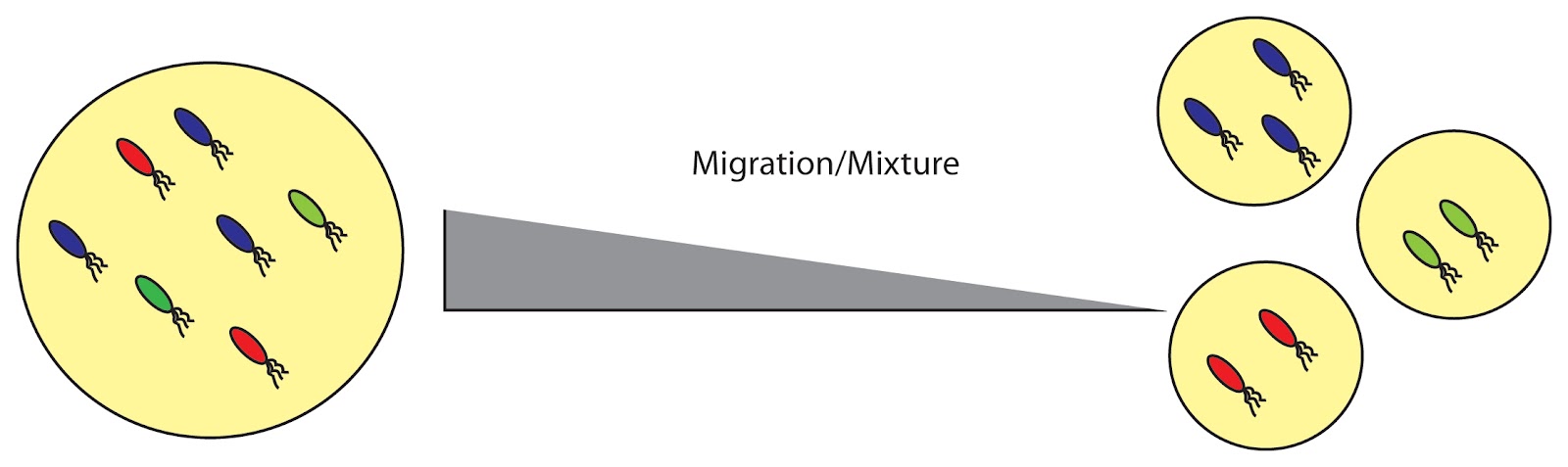
So how do we begin to measure microbial populations? I don't think it's been done yet, (please feel free to correct me!), but the rise of metagenomics at least makes the necessary experiments possible. With these technologies we can measure genotypes over time (all genotypes, not just a culturable subsample). We can measure how genotypes affect frequencies of other genotypes, and interactions could be evidence of existing within the same population. We could measure parameters such as strength of genetic drift and selection over time and extrapolate back to get Ne. Of course we then get into the issue of species level interactions, but I'll definitely save that for another time.
Thoughts?
Wednesday, June 27, 2012
And so it begins...
I’ve been thinking about starting a blog for a while, but it always seemed like there was too much activation energy involved. I guess that makes Jonathan Eisen my catalyst. I’ve seen what has taken place on Tree of Life (and even participated) as well as a variety of other terrific sites and have gained many insights (list of microbiology related blogs here). Communication with other researchers and the general public is increasingly taking place through social media, and I've been very happy with the types of interactions I've had on twitter. I hope this blog will be useful and fun for some people other than myself, but we’ll see. In the very least it provides a forum to crystalize thoughts so that, even if no one else sees this, I can look back and know where my brain was at.
What’s the content going to be? My research is focused on microbial evolution and my lab at the University of Arizona started a year and a half ago. Given that, I’m probably going to use this blog to give some insight into what life in the lab is like while fully acknowledging that there are a variety of other blogs out there with the very same intent. I love research and I love the thrill of figuring out new things, and will try to relate my research experiences (good and bad). I’m also a fan of arguing during journal clubs. While I was part of very lively journal clubs in grad school, my experiences since have left me wanting a bit. My hope is that I can use this as a forum start some good conversations about microbiology and evolution. Lastly, I really love a good history of science story and blogs provide a unique forum to tell such tales. In the very least I’m going to write about how some of my own papers happened, trials tribulations and all. Hopefully I can inspire some others to contribute as well. Look at this as an experiment, maybe it will work and maybe it wont, but I hope to learn something in the process.
Subscribe to:
Posts (Atom)