My new review in Trends in Ecology and Evolution went live last week. This paper, while not really experimental, took a little bit of a circuitous route and a bit of luck. For all of you out there sitting on ideas for reviews/opinions but not knowing how to get these published, here's how it happened (with a bit of philosophy of how to be a researcher thrown in).
One of the important skills I first learned in grad school was how to delve headfirst into a completely new topic and figure out the salient and relevant points. Sure, you might get sent a paper of interest by a colleague or have one picked for journal club, and these are great for getting a cliff's notes version of an area of research, but to understand a topic you need to know the background. One of the best ways for me to figure out the background (i.e why is the question interesting, what has been done in the past, what are directions for future research) was to find a review article and use that to point towards new references. These references can be found in the articles themselves, but its also quite helpful to search for other articles that cite the review. It doesn't even have to be a new article, because the rabbit hole of references eventually leads to the present day.
Somewhere along the way I developed a soft spot for the Trends family of journals (I know...Elsevier is evil...fully acknowledged, Frontiers is turning into a great place for reviews in the future). Trends articles were clear, concise, opinionated, and a great foothold for jumping into new areas. While there are other equally great resources for reviews like the Annual Reviews family, Bioessays, MMBR, etc...I set it as a goal early on in grad school to publish a first author article in Trends.
There are two different paths to write a Trends article: 1) you can receive an invitation from one of the editors or 2) you can submit a short pre-proposal and see if an Editor likes the idea. I was part of an invited review once, but this new article was the product of submitting and resubmitting pre-proposals.
When I started my lab I wanted to break away from what my postdoctoral advisor (Jeff Dangl) is known for and get back to my roots in microbial evolution. Sure, I still do a lot of phytopathology work in my lab, but I try to do this in the context of understanding how microbial pathogens evolve and adapt to new environments. This strategy has its positives and negatives, but long story short I found myself writing grant applications where the first page was devoted to explaining why the questions I was asking were interesting and important. This was too much space devoted to justifying my questions. Part of this is my lack of grant writing experiences, but part of this was that I felt I had to explain why I was asking the questions I was asking because, while there were many other articles preceding my ideas, there was no article (*as far as I can tell, point me towards these if they exist and I missed them please!) that laid these questions out in the context I was thinking about. I was simply using too many words to say something that could be easier said in a long review article and then cited.
My first stab at getting this review published was a pre-proposal to Trends in Microbiology. Obviously, this didn't work. I took a little bit of time, re-jiggered the ideas and foci, and submitted another pre-proposal to Trends in Ecology and Evolution (TREE). This time a very kind editor thought enough of what I wrote to give me a chance at a full article. I hadn't actually written the article yet, but the ideas were circulating in my head and most of the references I ended up using were gleaned from iterations of grants I was writing. I had about 3 months to write the full article, which is both plenty of time and not close to enough time, but I was able to pull together a draft and circulate it amongst my colleagues before submitting for formal peer review. This piece actually started out as an opinion, simply because I wasn't really sure if people were thinking about things the way I was. There is a feeling in science where you are both terrified and excited in the same moment. Either you are an idiot who is seeing things that other people have seen before and simply don't recognize this, or you are actually seeing things in a new way. The same feeling occurs when dealing with great new experimental results, except there is an extra option...either your experiment is 1) awesome sauce! 2) a trivial result that other people have seen before but which you haven't realized for one reason or another, or 3) a lab mistake.
I got the first reviews back and realized that I was onto something that other people were thinking about already (so...no controversial opinion needed), but that there was definitely a place for what I was writing. It's always difficult to read critique, but the reviewers actually did a great job pointing me towards other papers and helping me discover/emphasize other interesting research findings and directions. Specifically, the first iteration was too heavy on history and specifics and too light on the evolutionary implications of horizontal gene transfer. I made these changes, added a figure (in retrospect, should have changed the font and cut down on whitespace but I'm very happy with the information that it conveys), and sent it back in for a second review. This time it went through without a hitch, and I celebrated accordingly. Side note: I'm a slightly large person and have a tendency to break things in the house and the lab (My wife calls me Shrek). The sequence of the disruptive protein in figure 1 is an homage to my tendencies. A slightly less than subtle Easter egg, but I like trying to put those in my papers (there are plenty more subtle ones in other papers...). I'm also not alone in doing this.
The take home message is not to worry about whether something is good or not, just submit and see what happens. For a long time I was scared/worried about writing pre-proposals to the Trends editors, but it was a fairly seamless process once I went through it. You don't have to wait for the magical email invitation, just take the initiative and see if your idea flies. I'm writing my first grant after the review is out, and it's definitely much easier to write the background now. The last couple of paragraphs of the review also nicely set up some other manuscripts I'll be submitting this summer.
Thursday, May 30, 2013
Tuesday, May 7, 2013
Follow the biology (pt. 5) Season Finale
Yesterday I described assembling a reference genome for Pseudomonas stutzeri strain 28a24, in order to identify causative mutations for a brown colony/culture phenotype which spontaneously popped up while I was playing around with this strain. It's a pretty striking phenotype:

In addition to Illumina data for the reference parent strain, I also received sequencing reads for this brown colony phenotype strain as well as a couple of other independent derivatives of strain 28a24 (which will also be useful in this mutation hunt). There are a variety of programs to align reads to the draft genome assembly, like Bowtie2 and Maq. However, because it's a bit tricky to align reads back to draft genomes and because I'd like to quickly and visually be able to inspect the alignments, I'm going to use a program called Geneious for this post.
The first step is to import the draft assembly into Geneious, and in this case I've collapsed all of the contigs into one fasta read so that they are separated by 100 N's (that way I can tell what contigs I'm artificially collapsing from real scaffolded contigs). Next I import both trimmed Illumina paired read files, and perform a reference assembly vs. the draft genome. In Geneious it's pretty easy to extract the relevant variant information, like places where coverage is 0 (indicating a deletion) or small variants like single nucleotides or insertions/deletions. Here's what the output looks like:
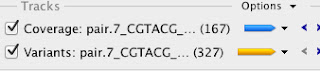
For the reads from the brown phenotype strain there are basically 167 regions where there are no reads mapped back to the draft genome. A quick bit of further inspection shows that these are all just places where I inserted the 100 N's to link together contigs. There are also 327 smaller variants that are backed up with sufficient coverage levels. My arbitrary threshold here was 10 reads per variant, but my coverage levels are way over that across the board (between 70 and 100x).
Here is where those other independent derivatives of the parent strain come into play. There are inevitably going to be assembly errors in the draft genome, and there are going to be places where reads are improperly mapped back to the draft genome. Aligning the independent non-brown isolate reads back against the draft genome and comparing lists of variants allows me to cull the list of variants I need to look more deeply at by disregarding variants shared by both. After this step I'm only left with 3 changes in the brown strain vs. the reference strain.
The first change is at position 2,184,445 in the draft genome, but remember that the number here is arbitrary because I've linked everything together. This variant is a deletion of a G in the brown genome.

Next step is to extract ~1000bp from around the variant and use blastx to give me an idea of what this protein is.
 Basically it's a chemotaxis signaling gene. Not the best candidate for the brown phenotype, but an indication that the brown strain probably isn't as motile as 28a24. Next variant up is at position 3,555,964. It's a G->T transversion potentially involved in choline transport...still not the best candidate for the brown phenotype.
Basically it's a chemotaxis signaling gene. Not the best candidate for the brown phenotype, but an indication that the brown strain probably isn't as motile as 28a24. Next variant up is at position 3,555,964. It's a G->T transversion potentially involved in choline transport...still not the best candidate for the brown phenotype.

The last variant is the most interesting. It's a T->G transversion in a gene that codes for homogentisate 1,2-dioxygenase (hgmA)


To illustrate the function of this gene, I'm going to pull up the tyrosine metabolism pathway from the KEGG server (hgmA is highlighted in red).

The function of HgmA is to convert homogentisate to 4-Maleyl acetoacetate. Innocuous enough and I'm no biochemist so in pre-Internet world I'd be somewhat lost right now. Luckily I have the power of google and knowledge of the brown phenotype so voila (LMGTFY). Apparently brown pigment accumulation in a wide variety of bacteria is due to a build up of homogentisic acid. There is even this paper in Pseudomonas putida. Oxidation of these compounds leads to quinoid derivatives, which spontaneously polymerize to yield melanin like things. Without doing any more genetics I'm pretty sure this is what I've been looking for. It's a glutamate to an aspartate change at position 338 in the protein sequence, a fairly innocuous change but which is (if this truly is the causal variant) in a very important part of the protein sequence. From here, if I were interested, I would clone the wild type version of hgmA and naturally transform it back into the brown variant of 28a24 to complement the mutation and demonstrate direct causality. I might also try and set up media without tyrosine to see if this strain is auxotrophic, which is a quality of other brown variants (see the P. putida paper above). For now I'll just leave it at that and move on to another interesting project that I can blog about.
In addition to Illumina data for the reference parent strain, I also received sequencing reads for this brown colony phenotype strain as well as a couple of other independent derivatives of strain 28a24 (which will also be useful in this mutation hunt). There are a variety of programs to align reads to the draft genome assembly, like Bowtie2 and Maq. However, because it's a bit tricky to align reads back to draft genomes and because I'd like to quickly and visually be able to inspect the alignments, I'm going to use a program called Geneious for this post.
The first step is to import the draft assembly into Geneious, and in this case I've collapsed all of the contigs into one fasta read so that they are separated by 100 N's (that way I can tell what contigs I'm artificially collapsing from real scaffolded contigs). Next I import both trimmed Illumina paired read files, and perform a reference assembly vs. the draft genome. In Geneious it's pretty easy to extract the relevant variant information, like places where coverage is 0 (indicating a deletion) or small variants like single nucleotides or insertions/deletions. Here's what the output looks like:
For the reads from the brown phenotype strain there are basically 167 regions where there are no reads mapped back to the draft genome. A quick bit of further inspection shows that these are all just places where I inserted the 100 N's to link together contigs. There are also 327 smaller variants that are backed up with sufficient coverage levels. My arbitrary threshold here was 10 reads per variant, but my coverage levels are way over that across the board (between 70 and 100x).
Here is where those other independent derivatives of the parent strain come into play. There are inevitably going to be assembly errors in the draft genome, and there are going to be places where reads are improperly mapped back to the draft genome. Aligning the independent non-brown isolate reads back against the draft genome and comparing lists of variants allows me to cull the list of variants I need to look more deeply at by disregarding variants shared by both. After this step I'm only left with 3 changes in the brown strain vs. the reference strain.
The first change is at position 2,184,445 in the draft genome, but remember that the number here is arbitrary because I've linked everything together. This variant is a deletion of a G in the brown genome.
Next step is to extract ~1000bp from around the variant and use blastx to give me an idea of what this protein is.
The last variant is the most interesting. It's a T->G transversion in a gene that codes for homogentisate 1,2-dioxygenase (hgmA)
To illustrate the function of this gene, I'm going to pull up the tyrosine metabolism pathway from the KEGG server (hgmA is highlighted in red).
The function of HgmA is to convert homogentisate to 4-Maleyl acetoacetate. Innocuous enough and I'm no biochemist so in pre-Internet world I'd be somewhat lost right now. Luckily I have the power of google and knowledge of the brown phenotype so voila (LMGTFY). Apparently brown pigment accumulation in a wide variety of bacteria is due to a build up of homogentisic acid. There is even this paper in Pseudomonas putida. Oxidation of these compounds leads to quinoid derivatives, which spontaneously polymerize to yield melanin like things. Without doing any more genetics I'm pretty sure this is what I've been looking for. It's a glutamate to an aspartate change at position 338 in the protein sequence, a fairly innocuous change but which is (if this truly is the causal variant) in a very important part of the protein sequence. From here, if I were interested, I would clone the wild type version of hgmA and naturally transform it back into the brown variant of 28a24 to complement the mutation and demonstrate direct causality. I might also try and set up media without tyrosine to see if this strain is auxotrophic, which is a quality of other brown variants (see the P. putida paper above). For now I'll just leave it at that and move on to another interesting project that I can blog about.
Monday, May 6, 2013
Follow the biology (pt. 4). Now we are getting somewhere...
First off, apologies on the complete lack of updates. In all honesty, there hasn't been much going on with this project since last summer but now I'm finally at the point of figuring out what kinds of genes are behind this mysterious brown colony phenotype in P. stutzeri.
Picking up where I left off , I've been through many unsuccessful attempts to disrupt the brown phenotype with transposon mutagenesis. The other straightforward option for figuring out the genetic basis for this effect is to sequence the whole genome of the mutant strain and find differences between the mutant and the wild type. Luckily this is 2013 and is easily possible. Confession time, while I know my way around genome scale data and can handle these type of analyses, I am by no means a full fledged bioinformaticist. I'm a microbial geneticist that can run command line unix programs and program a little bit in perl and python, out of necessity. If you have advice or ideas or a better way to carry out the analyses I'm going to describe below, please leave a comment or contact me outside of the blog. I'm always up for learning new and better ways to analyze data!
Long story short, I decided to sequence a variety of genomes using Illumina 100bp PE libraries on a HiSeq. For a single bacterial genome one lane of Illumina HiSeq is complete and utter overkill, when you can multiplex and sequence 24 at a time it's still overkill but less so. In case you are wondering, my upper limit it 24 for this run because of money not because of lack of want. It's a blunt decision, but one of many cost-benefit types of decisions you have to weigh when running a lab on a time dependent budget.
Before I even start to look into the brown mutant genome (next post), the first thing that needs to be done is sequencing and assembly of the reference, "wild type" strain. The bacteria I'm working with here is Pseudomonas stutzeri, specifically strain 28a24 from this paper by Johannes Sikorski. I acquired a murder (not the right collective noun, but any microbiologist can sympathize) of strains from Johannes a few years ago because one of my interests is on the evolutionary effects of natural transformation in natural populations. At the time I had just started a postdoc in Jeff Dangl's lab working with P. syringae, saw the power of Pseudomonads as a system, and wanted to get my hands on naturally transformable strains. Didn't quite know what I was going to do with them at the time, but since I started my lab in Tucson this 28a23 strain has come in very handy as an evolutionary model system.
I received my Illumina read files last week from our sequencing center, and began the slog that can be assembly. While bacterial genome assembly is going to get much easier and exact in the next 5 years (see here) right now working with Illumina reads is kind of like cooking in that you start with a base dish that is seasoned to flavor. My dish of choice for working with Illumina reads is SOAPdenovo. Why you may ask? Frankly, most of the short read assemblers perform equally on bacterial genomes for Illumina PE. Way back when I started using Velvet as an assembler, but over time became frustrated with the implementation. I can't quite recall when it happened, but it might have been when Illumina came out with their GAII platform and the computer cluster I was working with at the time didn't have enough memory to actually assemble genomes with Velvet. Regardless...I mean no slight to Daniel Zerbino and the EMBL team, but at that point I went with SOAPdenovo and have been working with this since due to what I'd like to euphemistically call "research momentum".
Every time I've performed assemblies with 100bp PE reads, the best kmer size for the assemblies always falls around 47 and so I always start around that number and modify as necessary (In case you are curious, helpful tutorials for how De Bruijn graphs work can be found here). One more thing to mention, these reads have already been trimmed for quality. The first thing I noticed with this recent batch of data was that there was A HUGE AMOUNT of it, even when split into 24 different samples. The thing about bacterial genome assemblies is that they start to crap out somewhere above 70 or 80x coverage, basically because depth and random errors confuse the assemblers. If I run the assembler including all of the data, this is the result:
2251 Contigs of 2251 are over bp -> 4674087 total bp
218 Contigs above 5000bp -> 1612110 total bp
30 Contigs above 10000 bp -> 352858 total bp
0 Contigs above 50000 bp -> 0 total bp
0 Contigs above 100,000 bp -> 0 total bp
0 Contigs above 200,000 bp -> 0 total bp
Mean Contig size = 2076.44913371835 bp
Largest Contig = 20517
The output is from a program Josie Reinhardt wrote when she was a grad student in Corbin Jones' lab (used again because of research momentum). The first line gives the total amount of contigs and scaffolds in the assembly, as well as the total assembly size. In this case, including all of the data yields 2251 total contigs, a total genome size of 4.67Mb, and a mean/largest contig of 2076/25,517bp. Not great, but I warned you about including everything.
Next, I'm going to lower the coverage. Judging by this assembly and other P. stutzeri genome sizes, 4.5Mb is right where this genome should be. For 70x or so coverage I am going to only want to include approximately 1.6 million reads from each PE file ( (4,500,000*70) / 200). When I run the assembly this time, the output gets significantly better:
467 Contigs of 467 are over bp -> 4695960 total bp
224 Contigs above 5000bp -> 4375593 total bp
158 Contigs above 10000 bp -> 3890697 total bp
11 Contigs above 50000 bp -> 709014 total bp
0 Contigs above 100,000 bp -> 0 total bp
0 Contigs above 200,000 bp -> 0 total bp
Mean Contig size = 10055.5888650964 bp
Largest Contig = 81785
I can show you more of this type of data with different amounts of coverage, but it's not going to change the outcome. The only other parameter to change a bit is kmer size. The figures above are for 47, but what happens if I use 49 with this reduced coverage?
382 Contigs of 382 are over bp -> 4694856 total bp
178 Contigs above 5000bp -> 4429753 total bp
130 Contigs above 10000 bp -> 4065927 total bp
18 Contigs above 50000 bp -> 1317727 total bp
2 Contigs above 100,000 bp -> 211768 total bp
0 Contigs above 200,000 bp -> 0 total bp
Mean Contig size = 12290.1989528796 bp
Largest Contig = 110167
Even better (and the best assemblies I've gotten so far). Increasing kmer size to 51 makes it incrementally worse:
479 Contigs of 479 are over bp -> 4633694 total bp
164 Contigs above 5000bp -> 4361629 total bp
124 Contigs above 10000 bp -> 4057109 total bp
19 Contigs above 50000 bp -> 1389519 total bp
1 Contigs above 100,000 bp -> 120161 total bp
0 Contigs above 200,000 bp -> 0 total bp
Mean Contig size = 9673.68267223382 bp
Largest Contig = 120161
Picking up where I left off , I've been through many unsuccessful attempts to disrupt the brown phenotype with transposon mutagenesis. The other straightforward option for figuring out the genetic basis for this effect is to sequence the whole genome of the mutant strain and find differences between the mutant and the wild type. Luckily this is 2013 and is easily possible. Confession time, while I know my way around genome scale data and can handle these type of analyses, I am by no means a full fledged bioinformaticist. I'm a microbial geneticist that can run command line unix programs and program a little bit in perl and python, out of necessity. If you have advice or ideas or a better way to carry out the analyses I'm going to describe below, please leave a comment or contact me outside of the blog. I'm always up for learning new and better ways to analyze data!
Long story short, I decided to sequence a variety of genomes using Illumina 100bp PE libraries on a HiSeq. For a single bacterial genome one lane of Illumina HiSeq is complete and utter overkill, when you can multiplex and sequence 24 at a time it's still overkill but less so. In case you are wondering, my upper limit it 24 for this run because of money not because of lack of want. It's a blunt decision, but one of many cost-benefit types of decisions you have to weigh when running a lab on a time dependent budget.
Before I even start to look into the brown mutant genome (next post), the first thing that needs to be done is sequencing and assembly of the reference, "wild type" strain. The bacteria I'm working with here is Pseudomonas stutzeri, specifically strain 28a24 from this paper by Johannes Sikorski. I acquired a murder (not the right collective noun, but any microbiologist can sympathize) of strains from Johannes a few years ago because one of my interests is on the evolutionary effects of natural transformation in natural populations. At the time I had just started a postdoc in Jeff Dangl's lab working with P. syringae, saw the power of Pseudomonads as a system, and wanted to get my hands on naturally transformable strains. Didn't quite know what I was going to do with them at the time, but since I started my lab in Tucson this 28a23 strain has come in very handy as an evolutionary model system.
I received my Illumina read files last week from our sequencing center, and began the slog that can be assembly. While bacterial genome assembly is going to get much easier and exact in the next 5 years (see here) right now working with Illumina reads is kind of like cooking in that you start with a base dish that is seasoned to flavor. My dish of choice for working with Illumina reads is SOAPdenovo. Why you may ask? Frankly, most of the short read assemblers perform equally on bacterial genomes for Illumina PE. Way back when I started using Velvet as an assembler, but over time became frustrated with the implementation. I can't quite recall when it happened, but it might have been when Illumina came out with their GAII platform and the computer cluster I was working with at the time didn't have enough memory to actually assemble genomes with Velvet. Regardless...I mean no slight to Daniel Zerbino and the EMBL team, but at that point I went with SOAPdenovo and have been working with this since due to what I'd like to euphemistically call "research momentum".
Every time I've performed assemblies with 100bp PE reads, the best kmer size for the assemblies always falls around 47 and so I always start around that number and modify as necessary (In case you are curious, helpful tutorials for how De Bruijn graphs work can be found here). One more thing to mention, these reads have already been trimmed for quality. The first thing I noticed with this recent batch of data was that there was A HUGE AMOUNT of it, even when split into 24 different samples. The thing about bacterial genome assemblies is that they start to crap out somewhere above 70 or 80x coverage, basically because depth and random errors confuse the assemblers. If I run the assembler including all of the data, this is the result:
2251 Contigs of 2251 are over bp -> 4674087 total bp
218 Contigs above 5000bp -> 1612110 total bp
30 Contigs above 10000 bp -> 352858 total bp
0 Contigs above 50000 bp -> 0 total bp
0 Contigs above 100,000 bp -> 0 total bp
0 Contigs above 200,000 bp -> 0 total bp
Mean Contig size = 2076.44913371835 bp
Largest Contig = 20517
The output is from a program Josie Reinhardt wrote when she was a grad student in Corbin Jones' lab (used again because of research momentum). The first line gives the total amount of contigs and scaffolds in the assembly, as well as the total assembly size. In this case, including all of the data yields 2251 total contigs, a total genome size of 4.67Mb, and a mean/largest contig of 2076/25,517bp. Not great, but I warned you about including everything.
Next, I'm going to lower the coverage. Judging by this assembly and other P. stutzeri genome sizes, 4.5Mb is right where this genome should be. For 70x or so coverage I am going to only want to include approximately 1.6 million reads from each PE file ( (4,500,000*70) / 200). When I run the assembly this time, the output gets significantly better:
467 Contigs of 467 are over bp -> 4695960 total bp
224 Contigs above 5000bp -> 4375593 total bp
158 Contigs above 10000 bp -> 3890697 total bp
11 Contigs above 50000 bp -> 709014 total bp
0 Contigs above 100,000 bp -> 0 total bp
0 Contigs above 200,000 bp -> 0 total bp
Mean Contig size = 10055.5888650964 bp
Largest Contig = 81785
I can show you more of this type of data with different amounts of coverage, but it's not going to change the outcome. The only other parameter to change a bit is kmer size. The figures above are for 47, but what happens if I use 49 with this reduced coverage?
382 Contigs of 382 are over bp -> 4694856 total bp
178 Contigs above 5000bp -> 4429753 total bp
130 Contigs above 10000 bp -> 4065927 total bp
18 Contigs above 50000 bp -> 1317727 total bp
2 Contigs above 100,000 bp -> 211768 total bp
0 Contigs above 200,000 bp -> 0 total bp
Mean Contig size = 12290.1989528796 bp
Largest Contig = 110167
Even better (and the best assemblies I've gotten so far). Increasing kmer size to 51 makes it incrementally worse:
479 Contigs of 479 are over bp -> 4633694 total bp
164 Contigs above 5000bp -> 4361629 total bp
124 Contigs above 10000 bp -> 4057109 total bp
19 Contigs above 50000 bp -> 1389519 total bp
1 Contigs above 100,000 bp -> 120161 total bp
0 Contigs above 200,000 bp -> 0 total bp
Mean Contig size = 9673.68267223382 bp
Largest Contig = 120161
Last question you might have, since there are other P. stutzeri genomes publicly available, is why not use those and carry out reference guided assembly? I haven't really looked into this much yet, but with some quick analyses it doesn't seem like these genomes are really that similar to one another (~85% nucleotide identity, aside from many presence/absence polymorphisms) so it's not that easy to actually line them up by nucleotide sequence. Maybe better when I've got protein sequences, but that's for another post.
So now I'm ready to compare the brown phenotype genome vs. this assembled draft of 28a24. I know what the answer is, but I'm going to leave that until the next post.
Subscribe to:
Comments (Atom)